File formats¶
Fasta¶
BaMM webserver accepts sequences in FASTA format. Only nucleotide sequences with the letters A, C, G, T, and N are accepted.
BaMM¶
The motif model in BaMM-format is a file with extension .ihbcp (inhomogeneous bamm conditional probability)
It stores the conditional probabilities of the BaMM model for each position. Motif positions are separated by a new line.
Here is an example of BaMM files for a 2nd order motif of length W:
Motif model (extension: .ihbcp)
P_1(A) P_1(C) P_1(G) P_1(T)
P_1(A|A) P_1(C|A) P_1(G|A) P_1(T|A) P_1(A|C) P_1(C|C) ... P_1(T|T)
P_1(A|AA) P_1(C|AA) P_1(G|AA) P_1(T|AA) P_1(A|AC) P_1(C|AC) ... P_1(T|TT)
P_2(A) P_2(C) P_2(G) P_2(T)
P_2(A|A) P_2(C|A) P_2(G|A) P_2(T|A) P_2(A|C) P_2(C|C) ... P_2(T|T)
P_2(A|AA) P_2(C|AA) P_2(G|AA) P_2(T|AA) P_2(A|AC) P_2(C|AC) ... P_2(T|TT)
...
P_W(A) P_W(C) P_W(G) P_W(T)
P_W(A|A) P_W(C|A) P_W(G|A) P_W(T|A) P_W(A|C) P_W(C|C) ... P_W(T|T)
P_W(A|AA) P_W(C|AA) P_W(G|AA) P_W(T|AA) P_W(A|AC) P_W(C|AC) ... P_W(T|TT)
Where P_W(A|CT) is the conditional probability of observing A at motif position W following the context CT.
Background model (extension: .hbcp)
P(A) P(C) P(G) P(T)
P(A|A) P(C|A) P(G|A) P(T|A) P(A|C) P(C|C) ... P(T|T)
P(A|AA) P(C|AA) P(G|AA) P(T|AA) P(A|AC) P(C|AC) ... P(T|TT)
Where P(A|CT) is the conditional probabilty of observing an A following a CT context. P is trained on the negative sequence set if available. If no negative sequences are provided, P is learnt on the positive set.
MEME¶
PWM models can be uploaded in meme MEME Versions lower than MEME version 4 have not been tested and are thus not recommended.
This is an example MEME file generated by PEnG-motif. Note that the lines below MOTIF are providing additional annotation and can vary between tools and databases.
MEME version 4
ALPHABET= ACGT
Background letter frequencies
A 0.25864 C 0.240258 G 0.241035 T 0.260067
MOTIF TGASTCATCSC
letter-probability matrix: alength= 4 w= 11 nsites= 32240 bg_prob= 0 opt_bg_order= 2 log(Pval)= -20070.6 zoops_score= 0.763 occur= 0.939
0.00000011 0.00000020 0.00000005 0.99999958
0.00000019 0.00000019 0.99973792 0.00026177
0.99776745 0.00222652 0.00000086 0.00000516
0.00043767 0.31039140 0.68885416 0.00031674
0.00000172 0.00001118 0.00000463 0.99998242
0.00015724 0.99983966 0.00000142 0.00000168
0.99997258 0.00000054 0.00002521 0.00000166
0.00000828 0.25723305 0.00413273 0.73862594
0.02208222 0.92982459 0.00702223 0.04107105
0.16592142 0.34808874 0.35102692 0.13496293
0.07382397 0.51519489 0.17385206 0.23712915
MOTIF ATTRTTTGTTTT
letter-probability matrix: alength= 4 w= 12 nsites= 13728 bg_prob= 0.0 opt_bg_order= 2 log(Pval)= -893.0211792 zoops_score= 0.252 occur= 0.621
0.68648666 0.00624365 0.03511349 0.27215624
0.27477601 0.00371415 0.06135688 0.66015303
0.00009623 0.00107756 0.00017856 0.99864769
0.56885940 0.00127072 0.42884308 0.00102682
0.00040802 0.00205148 0.00037785 0.99716270
0.00159969 0.00023089 0.00047653 0.99769294
0.00016588 0.00006246 0.01915511 0.98061651
0.24886248 0.01232569 0.70805818 0.03075366
0.00018377 0.14974646 0.01920011 0.83086962
0.08978166 0.01159330 0.00815281 0.89047223
0.00074780 0.00028864 0.00068021 0.99828333
0.27042452 0.00127012 0.01194946 0.71635598
Motif occurrence¶
We store motif occurences in a file with extension .occurrence.
Occurrence files have 7 columns:
- seq
- the sequence identifier in the uploaded fasta file
- length
- the length of the fasta sequence
- strand
- whether the motif was found on the positive (
+) or reverse complemented (-) strand. - start..end
- the relative position of the motif in the sequence
- pattern
- the nucleotide sequence of the motif in the sequence
- p-value
- the estimated p-value of the motif occurrence
- e-value
- the estimated e-value of the motif occurence
This is an example of an occurrence file:
seq length strand start..end pattern p-value e-value
>chr5:119672047-119672247 209 + 23..31 GGCAGCTGT 0.00045 0.225
>chr9:21950422-21950622 209 + 23..31 AGCAGCTGC 4.78e-05 0.0239
>chr7:6410115-6410315 209 + 101..109 GGCACCTGC 0.0001 0.0502
Motif evaluation¶
The motif evaluation scores are stored in a file with extension .bmscore.
*.bmscore files have 6 columns:
- TF
- base name of the sequence data file
- #
- number of the motif
- d_avrec
- data set AvRec score - a score indicating how well the motif can distinguish input sequences from articially generated sequences
- d_occur
- fraction of sequences with a motif in the data set setting (see explanation above)
- m_avrec
- motif AvRec score - a score indicating how well the motif can distinguish sequences with a motif from artificially generated sequences or input sequences without a motif.
- m_occur
- fraction of sequences with a motif in the input set
You can find a detailed definition and discussion of the AvRec score and the difference between dataset and AvRec and motif AvRec, in the webserver publication [KRG+18]
This is an example of an .bmscore file for a dataset with three motifs:
TF # d_avrec d_occur m_avrec m_occur
JUN_D 1 0.668 0.552 0.705 0.948
JUN_D 2 0.367 0.328 0.383 0.958
JUN_D 3 0.161 0.874 0.392 0.408
FAQ¶
- I think I found a bug, how can I make you aware?
- The best way is to file an issue in our github repository. Additionally you can write an email to bamm@mpibpc.mpg.de. In any case, please provide as much information as possible for us to reproduce the bug, e.g. the link to the result page.
- How long are the results available on the server?
- We guarantee that the results will be accessible via job id for at least 3 months.
- What is the maximum size of files I can upload?
You can upload files with up to 50 MiB in size.
For larger sequence files, you can either use our commandline tools, or run the webserver locally after adapting the
MAX_UPLOAD_FILE_SIZEconfiguration option.You can find detailed instructions in the README. in the webserver’s github repository.
How do I prepare my ChIP-seq data?¶
ChIP-seq produces regions in the genome that are bound by the factor of interest. The genome however is full of short repeats that due to their high occurrences and informativeness can easily overwhelm the signal of the true binding motif. Careful preprocessing can be crucial for optimizing the true binding motif.
Following pipeline has so far yielded good results for us.
- Use your favorite peak caller to obtain peaks from non-redunant bound regions
- Rank the sequences by the score obtained for each peak (e.g. q-Value)
- Extract fasta sequences centered on the peak regions of fixed length (e.g 201)
- Submit a fasta file with sequences from the highest ranked peaks (e.g. 5000)
Can I use the server with long sequences (>250bp)?¶
BaMMserver uses a ZOOPS model for learning and evaluating its higher-order models. That means all motifs are trained independently from each other and every sequence in the input file is considered to have either exactly one or no occurrence of the motif.
This setting is optimized for short sequences that are strongly enriched for the motif of interest, e.g. generated by CLIP-,ChIP- or SELEX-based methods. For longer sequences (e.g. scanning full promoter sequences), our ZOOPS model has limitations:
- Low complexity repeat sequences (e.g. ATATATATAT repeats) are abundant in genomes. Repeats will appear as strong motifs, despite having little biological significance for most questions.
- Our evaluation metric AvRec is based on how well the motif can distinguish input sequences from scrambled sequences. In our ZOOPS model only the best motif occurrence per sequence is used for classification. The longer the input sequences the higher the chance of finding a motif by chance.
- This has several implications:
- Despite of their strong enrichment, low complexity motifs are often biologically irrelevant.
- all but very long and informative motifs (often low complexity repeats!) score poorly in the AvRec benchmark.
- Following options are possible:
- Use only the seeding stage (
Manual seed selection, see De-novo motif discovery) which learns PWMs using a MOPS model. Skip the refinement of the seeds to BaMMs. - Chop long sequences up into multiple smaller sequences (e.g. 100bp) to get a more robust performance estimation, especially when only few sequences (<1000) are used (see also Can I use the server with very few sequences?).
- Use only the seeding stage (
Can I use the server with very few sequences?¶
You need to have at least 10 sequences - robust performance evaluation requires 100 or more sequences.
The higher-order motif refinement and the motif quality evaluation relies on the ZOOPS (zero or one occurrence per sequence) model. Very long sequences (>500) with more than one motif are best chopped up into smaller sequences before uploading to the higher-order refinement.
The seeding stage itself uses a MOOPS model. It is therefore possible to scan a handful of very long sequences for enriched PWMs. You can circumvent the minimum requirement of sequences for the seeding stage by adding extra sequences with the sequence ‘NNNNNNNNNNNN’. Please note that these seeds cannot be optimized to higher-order models due to the ZOOPS assumption.
How do I figure out whether my motif is biologically relevant?¶
Motif learners find enriched sequence motifs from the input data. However statistical significant motifs do not have to have to play a role in regulatory processes. De-novo motifs should be analyzed carefully - regulatory function should not be ascribed without further validation. We offer several ways to help validating the motifs:
Infer relevance from P-value distribution¶
By performing quality control and only selecting the 1000-5000 most strongly bound sequences, true motifs should be present in a significant amount of sequences. Have a look at the p-value statistic for calculating the motif AvRec (upper right plot in evaluation panel).
- There are mainly two things to ensure (see also Motif p-value distribution indicates the relevance of the motif):
- The p-value distribution should be skewed towards low p-values (The more uniform the less prevalance/information is in the motif)
- There should be a significant portion of area above the orange line (meaning that a significant portion of input sequences carry the motif).
If your motif does not pass the above criteria, try setting stricter cutoffs for selecting the sequences or shortening the sequences. If this does not help, the motif probably not relevant.
Warning
Using only very few input sequences will increase the noise on the p-value distribution and may make it hard to interpret the plot (see also Can I use the server with very few sequences?)
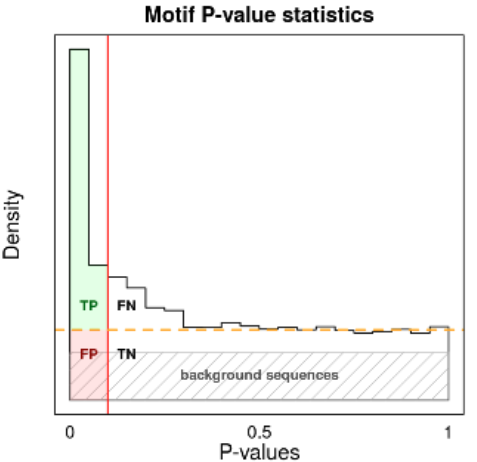
Motif p-value distribution indicates the relevance of the motif
Infer relevance from motif occurrences¶
When the sequences are generated from signal peaks (e.g. ChIP-seq), there is an additional information source available: when the sequences are extracted symmetrically around the peak, motifs should be enriched around the center.
The centered enrichment of motifs from ChIP-seq sequences can be appreciated in the figure below.
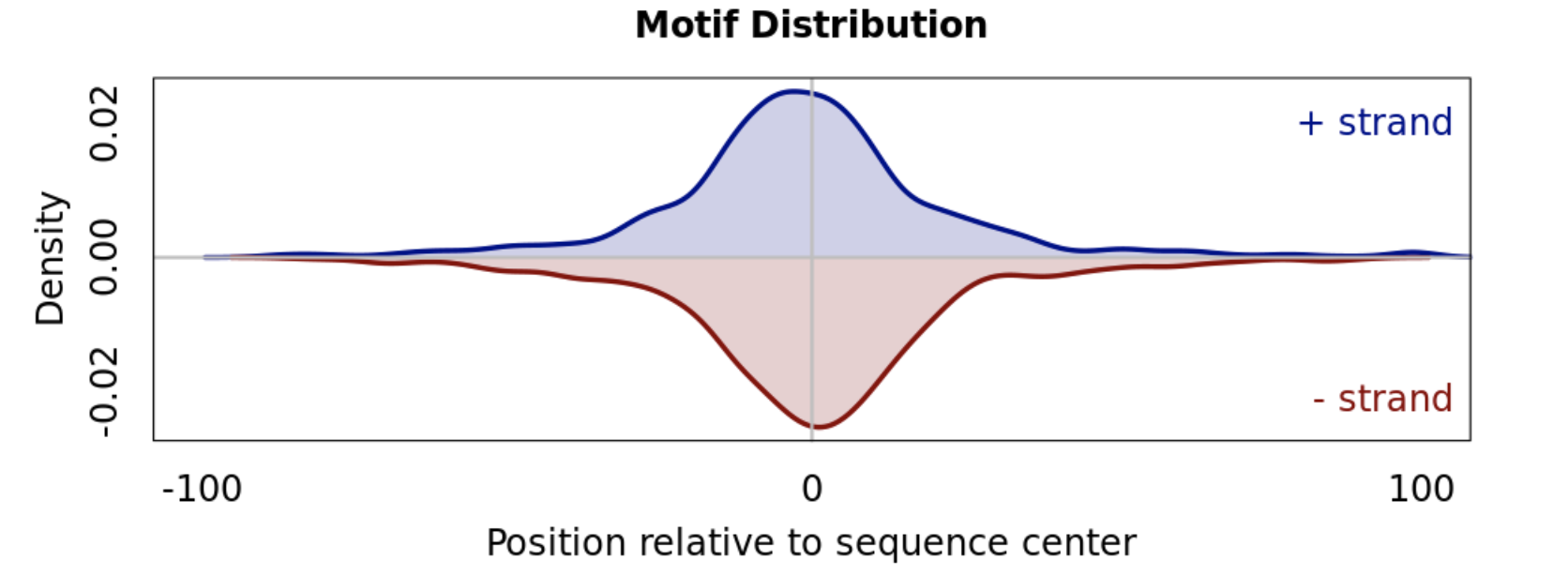
For sequences centered around peaks, motifs with uniform occurrence distributions are less likely to be of relevance.
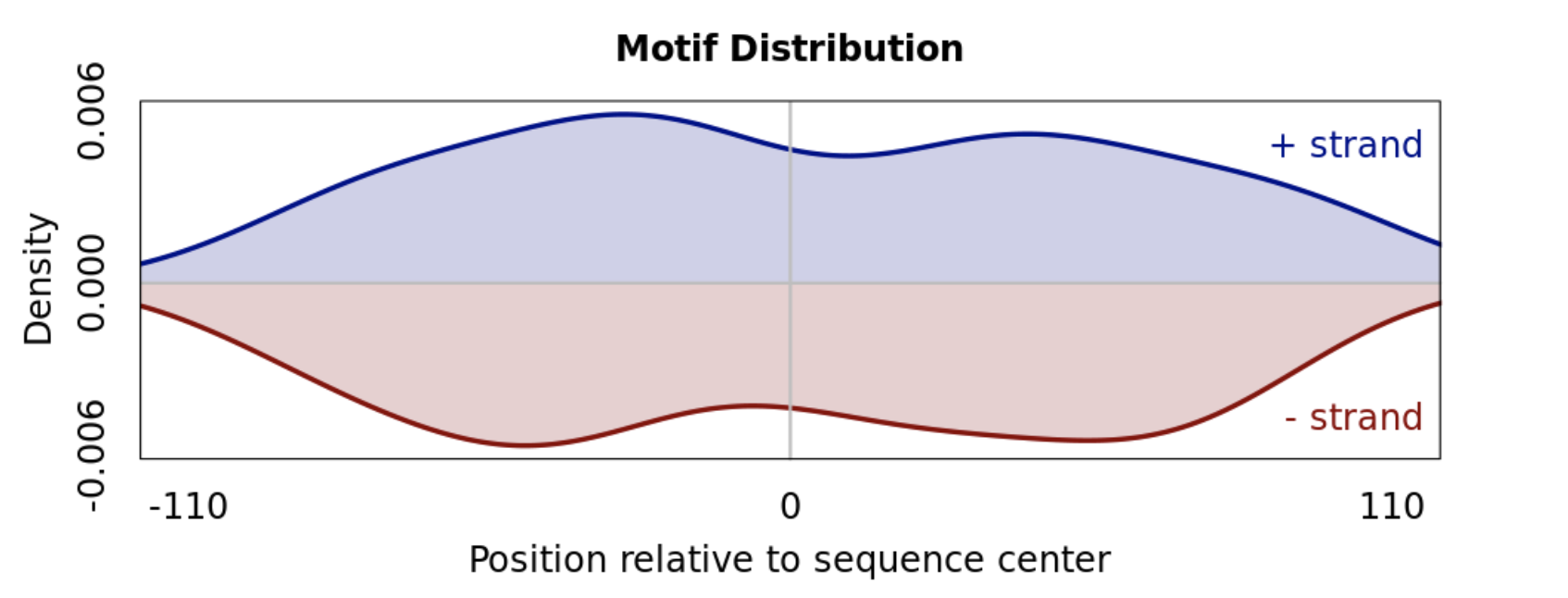
Infer relevance from motif complexity¶
Low complexity repeat regions are abundant in genomes. Always be careful when interpreting repeat motifs like ACACACAC or TATATATAT. The high repeat abundance and the high information content makes them easily reach statistical significance. They are especially prominent when the input sequences are long. The webserver will show a warning if the best scoring motif is a repeat motif.
Infer relevance from MMcompare annotation¶
Motif comparisons generated by our MMcompare tool can be used as a strong indicator that the motif is relevant and a good starting point for deeper investigation of the underlying biology.
Warning
Please do not forget to always be sceptical when assigning proteins and function to your discovered motifs. The motifs can originate from cofactors with strong binding motifs, or repetitive regions.

Where is the button to visualize in the genome browser?¶
The genome browser button

is only available if all fasta headers in your input file follow this format seqid:start-end with zero-based coordinates.
For hg38, an example fasta file would look like this:
>chr2:88600218-88600418
TGAAAGCAGATGGAGCTTTTCCTTGAGAGCCACAGAAGCAATATATGCATGCAGTTCAGGTACAGAGATGACATCACCCTTCACAATAGCATTACCTCACCCCCTAAGCATAGGAATGAGTCACCCGATAGTCAGCTGCAAATCTCTTGGTAGAAAAAAATGTAGGTTACGGTGATGCATTTTCACATCCCACTGATTTG
>chr7:37032027-37032227
TTTAAAAATATACTTGTTTGGCTTGATTCAGGCTGCTCCTCATTCCAGGCCTGCGTGAGTCATTGGAGAAACATCCTATTAGAGTGCACCCCTACTGATTGGCTTCCTTTGTATGTTCACGGTGACTCAGAAGAGATGACTCACAGTTCACGCTTATGACAAAAAGAACTTGCTCTCCCTTCCTTTTCATTACCCATGTT
[...]
You can generate an input file from genomic annotation files with the getfasta module of bedtools [QH10][Qui14].
Please refer to the documentation of bedtools for more details.
Miscellaneous¶
Glossary¶
- ZOOPS
- Zero or One Occurrence Per Sequence, describes the modeling assumption that input sequences can contain either no motif occurrence or at most one.
- MOPS
- Multiple Occurrences Per Sequences, describes the modeling assumption that input sequences can contain zero or multiple occurrences of a motif.
- AvRec
- Average Recall, evaluation metric used by the BaMMserver, for details see AvRec evaluation.
- PWM
- Position Weight Matrix, zeroth order motif model with independent contributions of each motif positions. See also PWMs on Wikipedia.
Using the commandline tools¶
The software for both the seeding stage (PEnG-motif) and the refinement stage (BaMMmotif) are available as standalone software packages under the GPL license. Please refer to the README files in the github repositories for more details how to use them.
Setting up the server locally¶
The source code of the server is open source and freely available under the AGPL license. If you intend setting up the server on your own computer, you can find a detailed description in the webserver’s README.
Citing BaMM webserver¶
If you are using BaMM webserver in your research, please cite our webserver [KRG+18] and BaMMmotif [SSoding16] papers, if applicable.